Très souvent à l’origine de calculs rénaux chez l’Homme, l’hyperoxalurie primitive est une pathologie génétique rare. Lorsque celle-ci apparaît, elle doit faire l’objet d’un diagnostic génétique précoce. En effet, le diagnostic précoce est nécessaire, car il pourra permettre une prise en charge systématique de la maladie. Ainsi, les complications de la maladie, en particulier la destruction des reins, pourra être retardée. Quelles sont les manifestations de l’hyperoxalurie primitive ? Quels sont les facteurs à l’origine de cette maladie et comment la traiter efficacement ?
Définition de l’hyperoxalurie primitive
L’hyperoxalurie primitive désigne un trouble du métabolisme du glyoxylate. Elle se caractérise par une surproduction d’oxalate, laquelle est à l’origine d’une insuffisance rénale, de calculs rénaux, d’une oxalose systémique et d’une néphrocalcinose.
En effet, l’oxalate est un déchet issu du métabolisme hépatique. Une fois formé, il est évacué de l’organisme par le biais des reins. Dans les urines, il s’associe au calcium, formant ainsi l’oxalate de calcium. Lorsque ce dernier est éliminé excessivement dans l’urine, on parle alors d’hyperoxalurie. À partir du moment où l’évacuation excessive de l’oxalate de calcium précède un phénomène d’ordre génétique, elle est dite primitive, d’où l’appellation hyperoxalurie primitive.
Par ailleurs, l’hyperoxalurie primitive est subdivisée en plusieurs types.
Types d’hyperoxalurie primitive
Jusqu’à preuve du contraire, on distingue exactement trois types d’hyperoxalurie primitive. Il s’agit de :
- L’hyperoxalurie primitive de type 1 ;
- L’hyperoxalurie primitive de type 2 ;
- L’hyperoxalurie primitive de type 3.
En effet, de toutes ces formes, l’hyperoxalurie primitive de type 1 est non seulement la plus courante, mais aussi la plus sévère. Elle peut occasionner une nécrose des reins et une oxalose (accumulation d’oxalate dans tous les organes et tissus sains).
Prévalence de l’hyperoxalurie primitive
L’hyperoxalurie primitive de type 1 est certes, la plus fréquente forme d’hyperoxalurie primitive, mais elle demeure une pathologie rare. En France, par exemple, on estime qu’elle touche 01 individu sur 110000 naissances. Actuellement, le nombre de personnes atteintes par cette maladie, dans ce pays de l’Europe, est évalué à 150 approximativement. On la retrouve un peu plus dans les pays où le mariage consanguin est courant. Dans ces régions, elle est d’ailleurs l’une des principales causes d’insuffisance rénale chez les plus petits.
Lorsqu’un individu, en particulier un enfant, est porteur d’une lithiase urinaire, il faut directement suspecter l’hyperoxalurie primitive, car celle-ci apparaît le plus souvent, très tôt, autrement dit, dès le début de la vie.
Parlant des hyperoxaluries de type 2 et 3, elles sont beaucoup plus rares et leur prévalence n’a pas réellement encore été estimée.
Causes et modes de transmission de l’hyperoxalurie primitive
Tous les types d’hyperoxalurie primitive ont un mode de transmission autosomique récessif. Toutefois, les gènes impliqués sont différents et sont au nombre de trois. Cependant, l’hyperoxalurie primitive ne survient qu’à partir du moment où les deux exemplaires du gène impliqué ont en leur possession des variants pathogènes, venant respectivement du père et de la mère. Étant donné que ces derniers portent chacun un variant pathogène, alors ils ne contracteront pas l’affection.
La survenue d’une hyperoxalurie primitive de type 1 est liée au déficit d’alanine glyoxylateaminotranférase. Il s’agit d’une enzyme hépatique, dont l’absence entrave le processus de transformation du glyoxylate en glycine, conduisant ainsi à une surproduction d’oxalate de calcium dans les urines. À ce niveau, il devient insoluble et entraîne la formation de calculs au niveau des voies urinaires, lesquels viennent se poser dans les reins, sous forme de cristaux. Par ailleurs, lorsque les reins ne parviennent plus à assurer leur fonction d’élimination, ces cristaux vont se poser au niveau des autres organes et tissus, entraînant donc une oxalose.
L’hyperoxalurie primitive de type 2 quant à elle, est provoquée par un manque de glyoxylate–hydroxy pyruvate réductase. Elle peut occasionner une oxalose ainsi qu’une insuffisance rénale. Enfin, la dernière forme de cette maladie est engendrée par l’absence de l’enzyme hydroxy–2-cétoglutarate aldolase. Elle peut conduire à des lithiases récidivantes et à une insuffisance rénale.
Quelle que soit la forme d’hyperoxalurie primitive, sa cause se localise au niveau du foie, car c’est à ce niveau qu’est produit l’oxalate, en excès.
Symptômes de l’hyperoxalurie primitive
Le plus souvent, cette pathologie hépatique se révèle précocement. En effet, elle se manifeste déjà avant l’âge de 5 ans chez plus de 45% des patients et avant la vingtaine, chez plus de 85% d’entre eux. De façon générale, l’hyperoxalurie primitive se traduit par :
- Des douleurs abdominales ou lombaires (chez l’enfant, elles sont similaires à la colique néphrétique) ;
- Une lithiase rénale;
- La présence de sang dans les urines (hématurie) ;
- Une infection urinaire.
Lorsqu’un individu souffre de cette maladie, on retrouve dans ses voies urinaires ou dans son tissu rénal, un dépôt de calcaire. Ce qui explique d’ailleurs la survenue récidivante des calculs.
Chez moins de 50% des patients atteints d’hyperoxalurie primitive 1, l’affection se manifeste, lors de sa première année d’apparition, par une insuffisance rénale, de forme infantile. À l’examen échographique, ils ont les reins pratiquement calcifiés : on parle de néphrocalcinose diffuse.
Par contre, l’apparition et l’évolution des symptômes de la maladie, différent d’un individu atteint à un autre, même si ces derniers sont de la même famille.
Diagnostic de l’hyperoxalurie primitive
Les symptômes que manifeste le patient sont significatifs à ce niveau, car ils permettent de déceler l’origine de la maladie. D’abord, le médecin procède à une intervention urologique, en vue d’analyser les calculs. Une telle analyse doit se faire dans un laboratoire de biochimie spécialisé. Lorsque celle-ci révèle la présence d’oxalate de calcium monohydraté, alors une hyperoxalurie primitive est soupçonnée.
Si le patient ne possède pas de calculs, le médecin effectue alors une cristallurie. Elle consiste à rechercher la présence de cristaux dans les urines du patient. Cette recherche peut donner des résultats similaires à la première forme d’intervention, qui consiste à analyser les calculs du patient.
Cependant, la présence de néphrocalcinose ou de lithiase, à l’échographie, doit pousser le médecin à réaliser un dosage d’oxalurie sur l’échantillon d’urine prélevé chez le patient. Ce dosage va servir à étudier le rapport d’évacuation de la créatinine et de l’oxalate. Chez l’adulte, lorsque l’excrétion quotidienne d’oxalate est supérieure à 0,5 millimole, alors il peut s’agir d’une hyperoxalurie primitive. Chez l’enfant, les normes en termes d’oxalurie varient selon l’âge. Toutefois, si l’insuffisance rénale est à un stade très avancé, les dosages urinaires ne pourront malheureusement pas être interprétés.
Pour confirmer le diagnostic de la maladie, le médecin procède ensuite à la recherche du gène à l’origine de la mutation pathogène. Pour ce faire, il doit réaliser un prélèvement sanguin. La confirmation du diagnostic doit se faire dans un laboratoire de biologie moléculaire. Ce diagnostic génétique va également permettre de détecter l’existence d’individus asymptomatiques se trouvant dans la famille du patient.
Il arrive que, dans certains cas exceptionnels, le variant pathogène en cause ne soit pas identifié. Dans ce cas, le médecin réalise une biopsie du foie pour prélever des cellules hépatiques. Sur ces dernières, il met en évidence les gènes AGT, GRHPR et HOGA. Par ailleurs, un tel examen est rarement indiqué.
Toutefois, il est possible de réaliser un diagnostic génétique prénatal, avec le consentement des parents du futur nourrisson. Cette intervention nécessite le prélèvement de villosités choriales, lequel doit se faire entre la 10ème et la 12ème semaine de grossesse. L’examen n’est possible que si le médecin note la présence du gène AGTX dans la famille.
Organes atteints par l’hyperoxalurie primitive
Le caractère insoluble de l’oxalate de calcium dans les liquides biologiques, entraîne la formation de cristaux, lesquels se posent aux alentours des tubules rénaux. Cela provoque une néphrocalcinose, qui est à l’origine de :
- La destruction progressive des reins;
- La formation de calculs dans la vessie et les uretères;
- L’obstruction partielle, voire complète des voies urinaires.
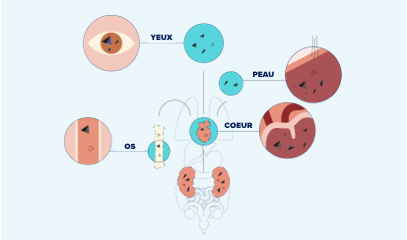
Lorsque l’insuffisance rénale est à un stade terminal, les reins ont du mal à éliminer les déchets. Une telle situation provoque une augmentation de l’oxalémie, entraînant ainsi le dépôt d’oxalate de calcium au niveau de la rétine, la moelle osseuse, la peau, des os, des artères, des articulations et du cœur.
Évolution de l’hyperoxalurie primitive
L’évolution de cette maladie hépatique est spécifique à chacune de ses différentes formes. Lorsqu’on prend le cas de l’hyperoxalurie primitive de type 1, elle peut occasionner une insuffisance rénale sévère ainsi qu’une destruction inéluctable des reins, en absence de prise en charge adaptée.
Aussi, les organes qui sont atteints par la pathologie, notamment le cœur, les os, la rétine, la peau ainsi que les artères, sont exposés à certaines complications. Par exemple, au niveau du cœur, le patient est assujetti au développement d’un trouble du rythme cardiaque. C’est pour cette raison que la prise en charge précoce de la maladie est nécessaire.
Parlant des deux autres types d’hyperoxalurie primitive, leurs complications sont habituellement moins sévères et elles ont une évolution plus ou moins lente.
Traitement de l’hyperoxalurie primitive
Dans le cas de l’hyperoxalurie primitive, le patient peut bénéficier d’une diversité de traitement, notamment un traitement préventif, un traitement urologique et une transplantation combinée foie–rein.
Traitement préventif
La prise en charge préventive vise à préserver le patient des complications de cette maladie hépatique. Pour ce faire, il faudra intervenir précocement, afin d’empêcher le dépôt de cristaux d’oxalate de sodium dans les reins. Cependant, le médecin devra au préalable échanger avec le patient sur les différentes mesures thérapeutiques envisageables.
L’option la plus fiable pour réduire la production d’oxalate consiste à faire usage de la vitamine B6. En effet, cette dernière intervient comme un cofacteur de l’enzyme en manque. Dans certains cas où l’on note la présence de l’enzyme, la vitamine B6 va intensifier l’activité de cette dernière et réduire considérablement la sécrétion d’oxalate. Chez moins de 50% des patients, l’usage de cette vitamine a contribué à la baisse de plus de 30% du taux d’oxalate, en seulement un mois.
Une autre forme de prise en charge consiste à administrer des bactéries au patient, en particulier les oxalobacterfomigenes. Lesquelles seront chargées d’éliminer le surplus d’oxalate, au niveau de l’intestin. Par ailleurs, il n’est pas certain que ce traitement soit si efficace, en raison de l’hyperproduction d’oxalate. Néanmoins, il peut être utilisé en complément de l’usage de la vitamine B6.
Il est aussi conseillé aux patients de suffisamment s’hydrater, afin de permettre une dilution maximale de l’urine. Toutefois, cette option n’est envisageable que si les reins assurent toujours efficacement leur fonction d’élimination. La quantité journalière d’eau à consommer est de 4 litres. Ainsi, l’urine sera permanemment et suffisamment diluée. Aussi, durant le sport ou lorsqu’il fait excessivement chaud, la prise de boissons peut être utile. Il faudrait ajouter qu’un arrêt de l’hyperhydratation peut occasionner l’apparition brusque d’oxalate dans les reins. Outre la voie orale, l’apport en eau peut se faire par voie veineuse.
Traitement urologique
Dans certains cas, le traitement médical seul n’est pas suffisant pour traiter les calculs déjà formés, d’où l’importance du traitement urologique. Ce dernier doit être réalisé par un expert urologue. Celui-ci devra privilégier un traitement endoscopique. Cependant, la pratique d’une chirurgie ouverte et d’une lithotrie extracorporelle est à proscrire.
Transplantation combinée foie-rein
Au fur et à mesure qu’évolue l’insuffisance rénale, cette double transplantation peut être nécessaire. Elle va empêcher une atteinte extra-rénale de l’oxalose systémique. C’est un traitement qui exige la prise d’un immunosuppresseur antirejet. Ce dernier sera utilisé pour toute la vie.
Une double transplantation peut être effectuée avec les organes d’un donneur décédé. En premier lieu, la greffe de foie sera réalisée. Elle doit être précédée d’une hépatectomie complète. Celle-ci va permettre une élimination de l’excès d’oxalate. Ensuite, la greffe rénale pourra être pratiquée, à condition que le stock d’oxalate soit éliminé. Cette forme de prise en charge nécessite la participation et l’intervention d’une équipe multidisciplinaire hautement qualifiée. La transplantation combinée foie–rein est réalisable chez l’enfant. En effet, ses résultats sont encourageants.
Surveillance de l’hyperoxalurie primitive
Un suivi du traitement préventif doit être réalisé. Son importance varie selon plusieurs critères, notamment l’âge du patient, la gravité de la maladie et son évolution. La surveillance doit prendre en compte :
- Les apports hydriques ;
- La fonction rénale ;
- La dilution des urines ;
- La diurèse.
La radiographie des voies urinaires, l’échographie et certains examens oculaires et cardiaques sont nécessaires pour apprécier l’absence de progression des dépôts d’oxalate. Pour cela, le traitement préventif doit être réalisé à la perfection.
En effet, lorsqu’il est bien réalisé, il empêche le dépôt d’oxalate dans les reins du patient traité précocement, et ralentit l’évolution des dépôts chez ceux traités tardivement. Il est donc d’une grande utilité. C’est son aboutissement qui donnera lieu à la surveillance de la maladie.



