La maladie ou le syndrome de Gilbert est une maladie génétique du foie provoquée par un déficit partiel d’élimination de la bilirubine. En effet, elle fait partie d’un groupe d’hyperbilirubinémie constitutionnelle, c’est-à-dire une augmentation du taux de bilirubine, la couleur primaire de la bile. Ce syndrome est génétique et est transmis à la descendance. Afin de cerner les contours de cette maladie, découvrez ici les facteurs fréquemment en cause, les symptômes, le diagnostic et le mode de traitement approprié.
Maladie de Gilbert : présentation
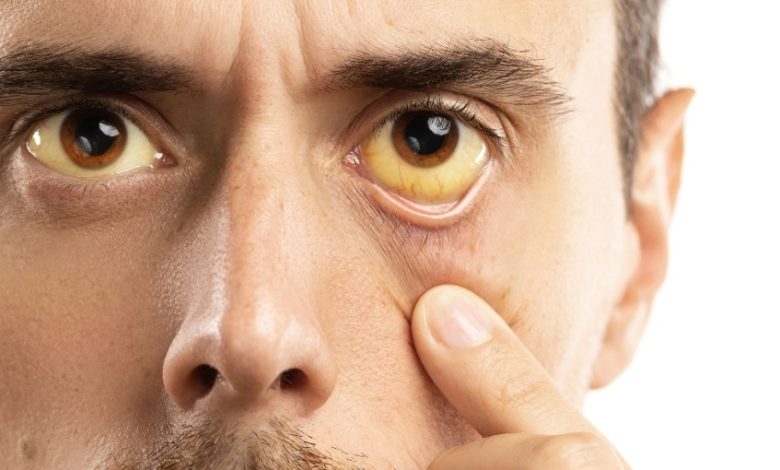
La maladie de Gilbert, également connue sous le nom de « syndrome de Gilbert », se caractérise par un niveau anormalement élevé de bilirubine chimique toxique dans le corps. De couleur jaune-orange, la bilirubine est créée à la décomposition des cellules sanguines.
Il faut noter que le taux de bilirubine fluctue considérablement chez les patients atteints de la maladie de Gilbert. Dans quelques rares cas, ce taux peut augmenter au point de provoquer une jaunisse ou un rougissement de la peau et des yeux.
Cette maladie est souvent diagnostiquée à l’adolescence. Cependant, il est possible qu’une personne subisse des épisodes d’hyperbilirubinémie. Ces épisodes ont fréquemment peu d’impact durable et surviennent lorsque l’organisme est en état de stress. Généralement, ils sont dus à des périodes de stress, une déshydratation prolongée, une maladie ou un exercice physique excessif.
En plus de la fatigue généralisée et chronique, les personnes atteintes de la maladie de Gilbert peuvent également présenter une gêne abdominale (problèmes de malabsorption, douleurs œsophagiennes, etc.).
Maladie de Gilbert : Processus métabolique de la bilirubine
Le principal sous-produit de la dégradation cellulaire du noyau héminique est la bilirubine, qui résulte principalement de la dégradation des globules rouges sénescents.
Suite à l’ouverture du cycle de l’hème par l’hème oxygénase, la biliverdine obtenue est réduite par la biliverdine réductase en bilirubine. La bilirubine non conjuguée étant très lipophile, elle doit être convertie en une forme hydrosoluble avant de pouvoir être éliminée de l’organisme.
La bilirubine, qui est liée à l’albumine, est transportée de son lieu de fabrication au foie où elle est conjuguée. Ce transport est crucial, car lorsque les capacités de liaison bilirubine-albumine sont dépassées, la bilirubine non conjuguée est libérée dans la circulation sanguine.
Il existe donc un risque important de lésions cellulaires, notamment au niveau de la structure cellulaire des yeux gris centraux. La bilirubine libre est capable de pénétrer toutes les membranes cellulaires en raison de sa haute lipophilie. Cependant, les cellules des centres gris sont plus sensibles à ses effets toxiques. Cela est probablement dû à un défaut de MRP1 qui n’est rien d’autre qu’un canal qui permet l’efflux actif de la bilirubine du cytosol vers le milieu extracellulaire.
Une fois dans la circulation hépatique, la bilirubine non conjuguée associée à l’albumine traverse la membrane hépatocellulaire, et ce par deux mécanismes. Elle peut emprunter la voie passive faisant intervenir la lipophilie et un gradient de concentration, soit la voie active utilisant partiellement le canal OATP2.
Maladie de Gilbert : les causes
Le syndrome de Gilbert résulte d’aberrations génétiques héréditaires transmises de génération en génération. Une carence en foie et la présence de bilirubine dans le sang sont les conséquences d’une erreur dans le codage de certains gènes.
En effet, lorsque les cellules sanguines atteignent leur maturité (après 120 jours), le processus biologique naturel de séparation de l’hémoglobine de la bilirubine a lieu. L’hémoglobine est le pigment sanguin rouge qui permet à l’oxygène d’être distribué dans tout le corps. De plus, le foie peut transformer ce produit chimique en une forme soluble dans l’eau.
En outre, la forme soluble de la bilirubine lui permet de pénétrer dans la bile puis d’être éliminée dans les urines ou les sécrétions sexuelles. Le foie d’une personne atteinte du syndrome de Gilbert ne peut plus rendre la bilirubine soluble. Dès lors, son transit par la bile n’est plus envisageable. Ainsi, ce médicament, qui n’a pas été éliminé, semble plus facile à trouver dans le sang.
Par ailleurs, il n’y a actuellement aucun facteur de risque connu pour la maladie de Gilbert au-delà du facteur génétique. Toutefois, tout le monde est susceptible de développer cette maladie. Cette dernière est transmise génétiquement et il existe un risque croissant qu’elle se développe également dans la famille. De plus, les femmes seraient plus susceptibles de contracter le mal.
Maladie de Gilbert : Symptômes
La maladie de Gilbert est souvent asymptomatique. Bien que les symptômes soient perceptibles, ils sont surtout représentés par un ictère à la surface de la peau et des conjonctives (yeux pâles et visages internes plus pauvres). Les patients qui ont la maladie de Gilbert ont fréquemment des symptômes d’identification ainsi que des épisodes de balade.
Les signes cliniques sont le plus souvent associés à :
- un état de déshydratation,
- un manque d’appétit peut indiquer une maladie,
- une période prolongée de stress couplée à un entraînement physique rigoureux,
- une absence de sommeil.
Certains adolescents et adultes se plaignent occasionnellement de brûlures d’estomac et de nausées. Néanmoins, on ignore si ces symptômes sont liés à la maladie de Gilbert ou en résultent.
Cependant, les individus atteints de la maladie de Gilbert ont des capacités limitées à éliminer la bilirubine. Ils partagent la capacité de produire des hormones (glucagon dans le cas des jeûnes, adrénaline et noradrénaline dans les autres cas) qui stimulent la formation de bilirubine. En conséquence, la bilirubine augmente dans le sang, ce qui entraîne une hyperbilirubinémie.
Maladie de Gilbert : Diagnostic
Du fait du caractère bénin de la cholémie familiale, cette affection n’est pas systématiquement recherchée. Toutefois, dans la grande majorité des cas, un diagnostic approfondi de la maladie de Gilbert est effectué par un examen systémique ou des tests sanguins.
Cependant, des tests sanguins peuvent être proposés aux parents des nourrissons présentant un ictère sévère ainsi qu’aux patients subissant certains traitements. En conséquence, certains médicaments sont dégradés par des mécanismes similaires à ceux utilisés pour éliminer la bilirubine. De plus ces médicaments peuvent devenir toxiques s’ils sont mal ou insuffisamment dégradés.
Par ailleurs, la maladie de Gilbert est diagnostiquée à l’aide d’une dose de bilirubine non conjuguée dans le sang pour mettre en évidence un cas isolé d’hyperbilirubinémie non conjuguée. Afin d’écarter le diagnostic d’hémolyse, le médecin prescrira également une numération de formule sanguine. Cette maladie du sang est plus grave et s’associe à une hyper-destruction des globules rouges.
Maladie de Gilbert : Manifestation clinicobiologique
Le principal symptôme du syndrome de Gilbert est une hyperbilirubinémie légère et fluctuante principalement non conjuguée. Le diagnostic peut être posé lors de l’investigation d’un subictère ou à l’issue d’un examen régulier qui révèle une hyperbilirubinémie.
Cependant, la sévérité de l’hyperbilirubinémie peut augmenter au cours du mois ou lors d’épisodes infectieux. Le reste de l’examen clinique est normal, sauf à partir de l’ictère. Certaines personnes ressentent de légères douleurs à l’estomac ou des nausées pendant les poussées d’ictus sans qu’un lien clair avec le déficit soit établi.
Par ailleurs, lorsqu’un patient souffrant du syndrome de Gilbert subit des crises d’ictus, il peut être difficile de faire la distinction entre un déficit enzymatique et une circonstance spécifique. Ce qui provoque une augmentation de la production transitoire de bilirubine (hyperhémolyse). Par conséquent, aucun diagnostic de syndrome de Gilbert, moléculaire ou autre, ne doit exclure la possibilité d’une pathologie globulaire.
En effet, plus de 100 enfants atteints d’ictère néonatal sévère nécessitent la mise en place d’une photothérapie. Ces enfants présentent une proportion plus faible de l’anomalie moléculaire responsable du syndrome de Gilbert que la population générale. La maladie de Gilbert est complètement bénigne et n’a pas besoin d’être traitée.
Les perspectives sont bonnes, la mise en place d’un suivi médical n’est donc pas nécessaire. Au contraire, certaines précautions doivent être prises lors de l’administration de médicaments qui utilisent la même voie métabolique. Leur élimination peut être diminuée, ce qui augmentera le risque de surdosage. Ceci est particulièrement bien décrit pour le médicament antimitotique irinotécan.
Maladie de Gilbert : Traitement
Cette pathologie n’est généralement pas grave et ne requiert pas un traitement complexe. Avec les mêmes recommandations pour tous, le patient est libre de poursuivre le régime de son choix. Il est souvent nécessaire de revoir de son mode de vie au quotidien. De fait, il peut être crucial de :
- privilégier une alimentation équilibrée ;
- limiter la consommation d’alcool ;
- pratiquer une activité physique de façon régulière.
Néanmoins, le diagnostic de la maladie de Gilbert commence par une analyse de sang qui permet de mesurer les niveaux de bilirubine ainsi que la fonction du foie.
Dans certaines circonstances, un test génétique peut appuyer un diagnostic clinique. La maladie de Gilbert est un mal chronique qui affecte la personne qui en souffre pour le reste de sa vie. De plus, il n’existe pas de traitement spécifique. Les problèmes de santé liés à la maladie de Gilbert sont peu fréquents. Dans certains cas, les effets secondaires les plus importants sont liés à l’apparition de maladies hépatiques (du foie). Quoi qu’il en soit, vous devez consulter un médecin le plus rapidement possible dès que vous remarquez les symptômes sus-cités. Le professionnel saura vous accompagner pour réduire au mieux les effets de la pathologie.



